FAQs
The majority of the information on this page can be found in our review:
- Tales from an academic RNAi screening facility; FAQs. Brief Funct Genomics. 2011 10:227-37.
What is the HTS Facility?
The High-Throughput screening facility at the Francis Crick Institute was set up in 2007 in order to make genome-wide RNAi screening readily accessible to the 45 basic research groups in the London Research Institute (CRUK) each with their own diverse research interests. We specialize in genome-wide siRNA screens primarily in human and Drosophila cultured cells. We also carry out several small-scale siRNA screens and chemical compound screens.
Do you need to have a dedicated facility to screen?
In outline, the process of screening a siRNA library is very simple; all that is required is to introduce RNAi reagent into cells uniformly across a number of multiwell plates and record the outcome. The problems start with the scale and it is this that can put the technology beyond the reach of any one single researcher. In our case, a genome-wide collection of pooled siRNA oligonucleotides consists of 267, 96-well library plates and so a screen in triplicate consists of 801, 96-well plates. A triplicate screen is equivalent to a stack of 96-well plates 14m high, consists of almost 77 000 wells (ie 77 000 transfections or immunostainings, etc.) and any manipulation that takes 1 min/plate will equate to 13 h needed to process the entire screen. Despite these numbers (not large by Pharma screening standards but usually disconcerting enough for academic researchers), it is still entirely possible to transfect 800 plates in a working day with only modest automation and a minimum of variability. Given the costs of siRNA collections and the infrastructure required to conduct large-scale assays (let alone the time investment needed to establish just one screen) it makes economic sense to provide screening as some form of core or shared resource for a number of interested parties. Such a core enables the assembly of the biological, programming and bioinformatics expertise required in one location, ensures a degree of continuity across many screens and affords the opportunity to create a central repository of data for the wider community.
Why do researchers screen and what are their expectations?
The reasons why researchers want to perform an siRNA screen are perhaps obvious; a screen can provide fresh insight or a new perspective on a process or area of biology or identify more precisely a general activity that is believed to exist from other work (all of which can be achieved to some extent). However, the hope that a single screen will yield a definitive catalogue of all activities required for a phenotype or identify genes that act universally in all cellular settings is unlikely to be immediately satisfied. Why don't people screen? Even with easy access to screening resources there are a number of reasons why people may not want to screen (aside from the obvious reason that it is an inappropriate tool for their needs). Fears about the possible variability and non-reproducibility of screens as well as considerations of the return for the time/cost investment have an element of truth about them that we shall explore below.
Like all experiments, screens are, one way or another, based on a central thesis, i.e. that there exists an activity which when knocked down elicits phenotype X. One can go a stage further and be more explicit in stating the central hypothesis:
Although this may seem to be pedantry for its own sake, it does focus attention on the assumptions inherent in the assay design, the points where assays might show greatest variation and where answers might not be as universally applicable as first hoped. Like genetic screens in model organisms , an siRNA screen faithfully reports the answers to the specific question posed so if the results of the screen do not match expectations, it is always worth reconsidering the assumptions underlying the assay and screen.
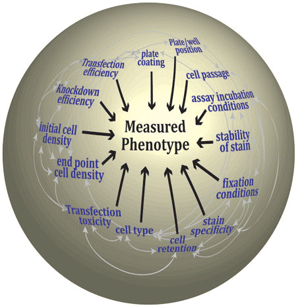
I have an idea for an assay, when can I screen?
While transferring an assay from a 'one pipette/ coverslip' environment to a 'multi-channel device/ multiwell plate' environment might seem to be a simple linear question of re-optimization for a smaller physical chamber, it is often less than straightforward. The many variables that make up an assay are interconnected such that alteration in one can have profound effects on the outcome of the assay (Figure 1). Although, some of the assay steps will be fixed (e.g. only one cell line or cell type or one class of assay readout is possible), the key here is that the development of the assay is an iterative process attempting to maximize the response while minimizing the experimental variation and identifying the level of tolerance of the phenotypic output to this variation. This process requires many test plates and pilot (small scale) studies that are best performed using the equipment that will be employed for the primary screen (especially if the assay is in 384-well plates) and moreover, requires constant interaction between data analysts and biologists to maximize the return of data. In our facility, much of our time is spent developing the assay with the researchers in a process that usually takes months rather than weeks but which ultimately equips us with an intimate knowledge of the assay system and the variables influencing the assay outcome.
What source of siRNA reagents should I use?
Anyone venturing into screening will be faced with the decision as to which siRNA collection to use; usually, the choice boils down to a commercial or homemade source. Despite anecdotal reports that siRNAs from supplier x are 'better' than those from supplier y (declarations often based on quite small sample sizes), we are unaware of any peer-reviewed literature clearly identifying one commercial supplier as preferable over another. Although all suppliers try to improve their design algorithm and bioinformatics filters that inform their siRNA designs, most collections will be out of date to some extent as a result of the rapid changes in genome annotation and ever expanding knowledge of small RNA biology. However, they are easy to manage and purchase (at a price).
What transfection reagent should I use?
One of the most frequently encountered and least discussed technical aspects is the toxicity of reagents used to transfer siRNA into cells. We have observed significant batch-to-batch variation among some reagents (and even 'within-batch' variation), reagent and cell type interactions, reagent-induced cell-specific morphology effects and reagent-induced assay readout interference. In our view, most reagents induce some form of stress in cells and how this stress manifests itself depends on the cell type and the assay conditions. We deal with this reagent problem empirically by iterative testing of reagent types and doses ideally in an assay mimicking the intended readout as far as possible and constantly monitor reagent performance while developing and perfecting the screen (Figure 2).
What cells should I use?
Cells are the central component in any assay and the component about which most assumptions are made with respect to their genotype, response and ability to serve as a model for a cell type in vivo, etc. Cell lines differ in their inherent ability to be transfected. We find that how cells are manipulated is crucial to the success of any assay and therefore try to enforce strictly standardized cell culture (including regular growth rate and contamination monitoring). Additionally, the age or passage of the culture per se influences the knockdown response and phenotypic outcome. Although not a universal practice, we try to complete genome-wide screens with one batch of cells rather than split into several batches over many weeks, as this can be equivalent to performing several similar but subtly distinct screens. Why might the maintenance and culture history of cells matter so much? It is clear that cells in culture are not uniform and the individual cells in any population display natural variation in cellular states and phenotypic response. It is probable that this variation contributes to some of the observed variation in response to siRNA challenge. Thus anything that might favour one cell state rather than another (like allowing cells to overgrow) can influence the measured phenotype. Cellular responses are also subject to plate positional, edge effects, particularly in 384-well and greater well formats that may not always reveal themselves as changes in viability. A common assay format is to compare the cellular responses to different incubation conditions (e.g. comparing two screens on the same cell line with and without drug) or to compare two related cell lines. For cell line comparisons, assumptions are made about the similarity or differences between them (either with respect to genotype or phenotypic response) and these assumptions may benefit from careful consideration. For example, recent technical advances are revealing extraordinary inter- and intra-tumour genotype and gene expression heterogeneity and presumably the same applies to the cell lines derived from them. So far, our experience suggests that screens aimed at such comparisons would benefit from more than one pair-wise comparison.
What readout should I use?
Ideally the readout of the assay should be limited only by the imagination of the researcher and what is measured should be as accurate and complete a representation of the desired phenotype as possible. It is in this area that a facility needs maximum technological flexibility. If the cost of the proposed screen readout is likely to be prohibitive or the assay protocol too technically challenging for the degree of automation available, most researchers are willing to consider alternative, substitute, measures which will be good enough (which requires that the system is sufficiently well characterized to recognize that a readout is good enough). Broadly speaking, we favour fixed-endpoint assays where the readout is captured on a cytometer or microscope rather than homogenous assays, as they afford the greatest opportunity for multiplexing and alternative data gathering. Indeed image analysis and collection of multiple parameters for each cell is an increasingly popular readout for many assays and may provide a robust approach to manage the biological variation seen in screens. Image based readouts also provide a facility with a wealth of bystander data (data not originally planned to be used by the researcher but which can be accumulated and amalgamated across screens to create an useful database, e.g. cell viability or cell shape).
What controls should I use?
Control siRNAs are usually included on every plate in a screen and can be used in different ways at different times in the screening process. We tend to use them as indicators of screen efficiency and for data curation in the primary screen and as tools for normalization in follow-up screens. Negative or null control siRNAs are often designed against non-mammalian proteins or are modified so that they do not enter into the RISC complex. We often find in analysing screen data that such controls cannot be assumed to occupy the central, medial position in the dataset as might be anticipated (since they have no effect) but instead, can often be skewed toward one end of the distribution. This is particularly evident in our hands when looking at viability assays; our interpretation has been that almost all siRNAs in the library can have some effect on viability and perhaps simply the engagement of the RISC complex is sufficient to retard cell growth to some extent in some lines. Often the point of the screen is to identify putative activities so positive control siRNAs (i.e. that can elicit the desired phenotype) may not be readily available. It is possible to develop an assay and prosecute a screening campaign in the absence of a positive control but very much harder to do so. It is also common practice to use an siRNA which produces a recognizable phenotype as a control to demonstrate that the transfection was effective.
How do I analyse the data?
Much work has gone into identifying appropriate methods to analyse RNAi data to compensate for assay bias and enrich for true hits by statistical and bioinformatics analysis. Since different assays can have distinct response characteristics there is unlikely to be only one best way to analyse the data. Certainly we would advise visualizing data graphically as a good way of identifying problems in a dataset. We also find it best to be ruthless and disregard data where there is clearly a problem (e.g. where the control siRNAs in a minority of plates are out of keeping with the rest of the dataset). We have quickly adapted methods to identify such problems and feed this information back to the screeners to modify future screening campaigns if necessary. It is our impression that strong hits or responses usually remain so whatever the type of analysis employed although hit classification for weaker effects can vary enormously depending on the technique employed. From the researchers' point of view, the important question is whether it is possible to employ some measures or analysis of the data that maximizes the return of biologically relevant hits or hits that will in some sense be true. Although there have been a number of additional bioinformatic filters and analyses employed to increase the frequency of selecting genuine biological hits from RNAi data , we are of the opinion that ultimately there is probably a limit to what statistics and analysis can provide given the degree of biological variation and pragmatically it might be best to be more generous with thresholds for defining hits and spend more time on further experiments.
How do I validate my hits?
The phrase 'hit validation' maybe an example where the choice of language can impact on the perception of screen data and the nature of the subsequent experimentation. Originally from the Pharma chemical screening environment (where it usually referred to the question 'Is the observed effect due to the chemical that I believe to be in that well'), 'validation' has begun to acquire a broader, less precise meaning. It does not imply biological validity (i.e. that the observation is true under all circumstances, or that loss of gene X alone causes the observed phenotype). Moreover, 'validation' isn't a process following a set of immutable rules with a defined end-point; like all research, the observation (the RNAi effect) will be constantly challenged as knowledge evolves and is only as 'valid' as the last experiment. The central question for siRNA screens is 'for a number of candidates from the primary screen, what is the quickest way to become confident that the effects elicited by those reagents are largely or solely the result of the disappearance of the intended protein'. Any approach that increases the confidence would be a 'validation' strategy and perhaps the best such corroborative data would come from techniques other than RNAi, e.g. mouse or zebrafish mutants. Most users agree that one of the best approaches that is amenable to larger numbers of samples is rescue of the phenotype by expression of a siRNA resistant clone, although precise modulation of the expression levels of the rescuing clone is essential, but not straightforward, to achieve. Even more straightforward and by far the most popular start point for validation is to show that the effect is produced by more than one oligonucleotide sequence. Such approaches are not without their problems as alternative siRNA reagents with different sequences may be inherently more or less effective than the original siRNA and interpretation of data from these screens can be further complicated by the fact that the relationship between the extent of mRNA decrease, decrease in protein levels and phenotypic change might not be linear for all genes. Validation requires a combination of many different techniques and approaches often in multiwell plate format and it is best if assays designed to classify hits from the primary screen are developed in conjunction with the primary assay. In short, the end of the screen is the start of a very long journey.
What are the common artefacts?
The most well-known artefact is that of off-target effects, i.e. unintended silencing of some activity other than the intended target. The mechanisms by which an off-target effect can be produced are many fold and well-reviewedand often such effects become apparent only after extensive work. As knowledge about the biology of small and noncoding RNAs increases, it is to be hoped that reagent design can improve further. However, it is possible that no matter what design algorithms are used, siRNA will always be to some extent 'dirty' reagents, i.e. having effects additional to those for which they were designed. Image-based screens recording multiple parameters per cell might make it easier to identify a true phenotype from apparently similar phenotypes generated by off-target effects. There is an alternative, more positive view of off-target effects: any oligonucleotide eliciting a phenotype solely through an off-target effect is still a reagent that elicits the desired phenotype (and therefore may well be pursuing if the research requires it) but the precise mechanism of its action is unknown, although it probably involves RNA at some point. A less well-described assay artefact we have encountered is the effect of cell death on a number of different readouts primarily luciferase assays and fluorescent protein assays. We observe that RNAi reagents that cause significant cell death can also disproportionately increase the fluorescent or luminescent output of those cells such that the top hits contain an over-representation of siRNAs inducing cell death.
Are siRNA screens reliable and reproducible?
Increasing numbers of publications clearly attest to the fact that RNAi screening works and identifies novel activities that can be shown by methods other than RNAi to be true. However, our numerous 'corridor conversations' about rumoured screen artefacts or the inability of researchers to reproduce a published observation using similar reagents suggest that there is still some unease about how to interpret results from siRNA screens. The similarity between replicates within any one screen is usually high, i.e. screens are self-consistent. But the frequency with which primary hits can be reproduced in secondary screens is variable (a combination of genuine reproduction failure, identification of the effect as entirely an off-target effect and possibly differences in the analysis of data between genome-wide and smaller scale experiments). What might make an individual RNAi reagent fail to reproduce from one occasion to another or from one researcher to another? For any one cell, the phenotype resulting from a reduction in the level of a target protein probably depends on:
- the rate and extent of the reduction (and whether the phenotype responds linearly to reduction requires a threshold before an effect is observed or shows an alternative more complex dose dependency)
- the degree of functional redundancy of the protein and whether the protein is involved in multiple cellular processes
- any specific phenotypes elicited by the transfection process per se
- any contributing phenotypic effects of off-target actions of the RNAi reagent
Within a cell population, the many possible distinct cellular states that co-exist could determine the measured penetrance of the phenotype in that population. All of these aspects could contribute to the day-to-day variability of assays and might therefore contribute to the failure of some primary screen 'hits' to reproduce even in the hands of the same researcher. It would also suggest that hits surviving several rounds of screening are likely to be those showing the least complex of dependencies.
When screening data from different laboratories addressing either similar or apparently identical areas of biology are compared, the overlap can be very disappointing. If each screen provides a precise answer to the question posed, it might be that there are sufficient differences in assay design and execution to explain the poor overlap. In that sense all screen data are 'correct' it is just that the questions posed were different in detail. Such an interpretation therefore focuses attention on the nature of the information sought from a screen. Given the possibility for variability in the phenotypes elicited by individual siRNAs it is unlikely that any one screen will give a complete and full inventory of all of the activities involved in a biological process in either one cell line or across all cell lines. Such data is therefore probably best garnered by multiple screens possibly across many lines or with distinct complementary readouts.